
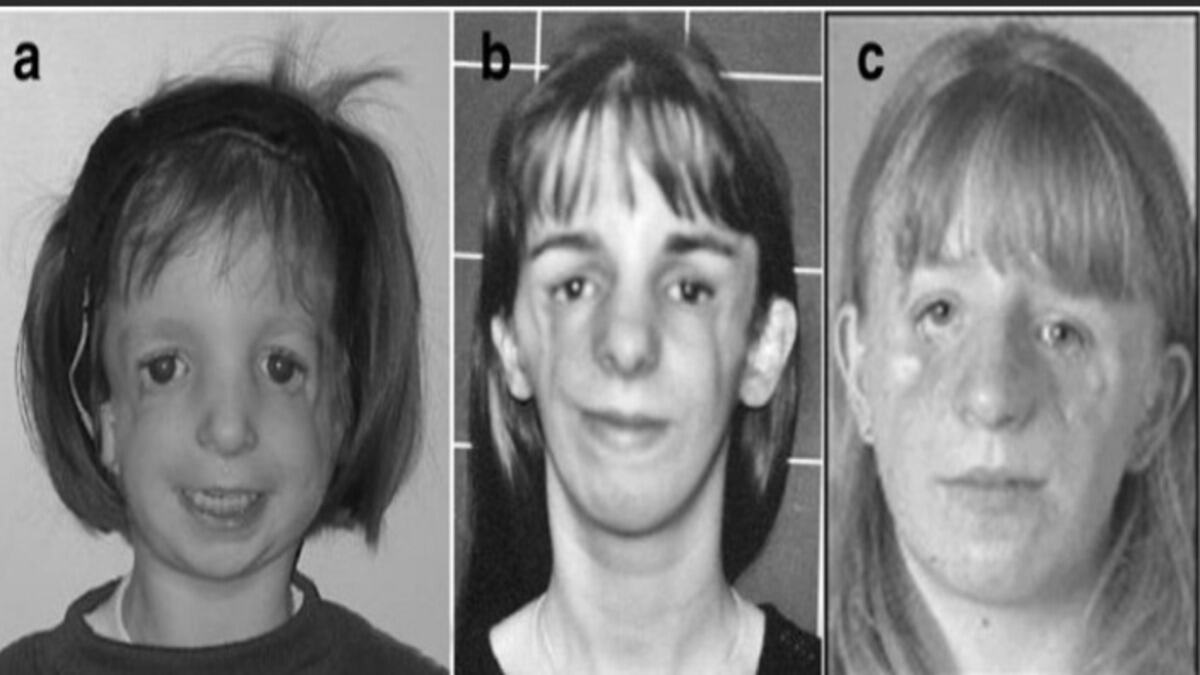
Taş adam sendromu, fibrodisplazi ossificans progresif (FOP) olarak da bilinmektedir . Bu nadir görülen genetik hastalık, vücudun bağ dokularının kemikleşmesine neden olur. Bağ dokusu, kasları, kemikleri ve organları birbirine bağlayan doku türüdür. FOP’lu kişilerde, bağ dokusu hasar gördüğünde veya iltihaplandığında, normalde kemik oluşumunun gerçekleşmediği bölgelerde bile kemikleşme meydana gelir.
Taş adam sendromu
FOP, dünya çapında yaklaşık 2.500 kişide görülen bir hastalıktır. Hastalığın nedeni, 2. kromozomun q (uzun) kolunun 23. ve 24. bölgelerindeki genlerdeki bir mutasyondur. Bu mutasyon, bağ dokusunun normal şekilde iyileşmesini sağlayan bir proteinin üretimini engeller.
FOP’un belirtileri genellikle erken çocukluk döneminde ortaya çıkar. Hastaların çoğu, doğumda veya birkaç ay içinde doğum lekesi veya şişlik gibi hafif belirtiler gösterir. Hastalık ilerledikçe, kemikleşme daha yaygın hale gelir ve kas hareketliliğini kısıtlamaktadır.
FOP, hastanın yaşam kalitesini önemli ölçüde etkilemektedir. Hastalar, kemikleşmenin neden olduğu hareket kısıtlılığı nedeniyle günlük aktivitelerini yerine getirmekte zorlanmaktadır. Ayrıca, kemikleşmenin neden olduğu ağrı ve rahatsızlık da hastaların yaşam kalitesini olumsuz yönde etkilemektdir.
FOP için henüz kesin bir tedavi yoktur. Ancak, hastaların ağrı ve rahatsızlıklarını azaltmaya yardımcı olmak için çeşitli tedaviler kullanılmaktadır. Bu tedaviler arasında fizik tedavi, ilaç tedavisi ve cerrahi müdahale yer alır.
FOP’lu bazı kişiler, hastalıklarını kabullenmeyi ve yaşam tarzlarını buna göre ayarlamayı öğrenmiştir.
FOP, nadir görülen ve ciddi bir hastalıktır. Ancak, hastalar doğru tedavi ve destek ile yaşamlarını mümkün olduğunca normal bir şekilde sürdürebilirler.
Taş adam sendromu : 17. Yüzyılda İlk Görülme
Taş adam sendromunun (FOP) ilk ne zaman görüldüğü kesin olarak bilinmemekle birlikte, en eski kayıtlar 17. yüzyıla kadar uzanmaktadır. Bu kayıtlarda, hastalığın belirtilerini gösteren birkaç kişiden bahsedilmektedir.
- yüzyılda, FOP’un daha ayrıntılı olarak tanımlanması için bazı çalışmalar yapılmıştır. 1896 yılında, İngiliz doktor John Caffey, hastalığın en yaygın belirtilerini tanımlayan bir makale yayınlamıştır.
- yüzyılda, FOP’un genetik temeli hakkındaki araştırmalar hız kazanmıştır. 1998 yılında, Amerikalı bilim adamları, FOP’a neden olan mutasyonu 2. kromozomda tanımlamışlardır.
FOP, nadir görülen bir hastalıktır. Dünyada yaklaşık 2.500 kişide görülmüştür. Hastalığın belirtileri genellikle erken çocukluk döneminde ortaya çıkar. Hastaların çoğu, doğumda veya birkaç ay içinde doğum lekesi veya şişlik gibi hafif belirtiler gösterir. Hastalık ilerledikçe, kemikleşme daha yaygın hale gelir ve kas hareketliliğini kısıtlamaktadır.
FOP, hastanın yaşam kalitesini önemli ölçüde etkilemektedir. Hastalar, kemikleşmenin neden olduğu hareket kısıtlılığı nedeniyle günlük aktivitelerini yerine getirmekte zorlanmaktadır. Ayrıca, kemikleşmenin neden olduğu ağrı ve rahatsızlık da hastaların yaşam kalitesini olumsuz yönde etkileyebilmektedir.
FOP için henüz kesin bir tedavi yoktur. Ancak, hastaların ağrı ve rahatsızlıklarını azaltmaya yardımcı olmak için çeşitli tedaviler kullanılmaktadır. Bu tedaviler arasında fizik tedavi, ilaç tedavisi ve cerrahi müdahale yer alır.
FOP’lu bazı kişiler, hastalıklarını kabullenmeyi ve yaşam tarzlarını buna göre ayarlamayı öğrenmiştir. Bu kişiler, engelliliklerine rağmen başarılı bir şekilde çalışmaktadır. Bunun sonucunda aile kurabilmektedir. Buna bağlı olarak sosyal hayatlarına devam edebilmektedir .

